
In-Fusion Cloning
Introduction
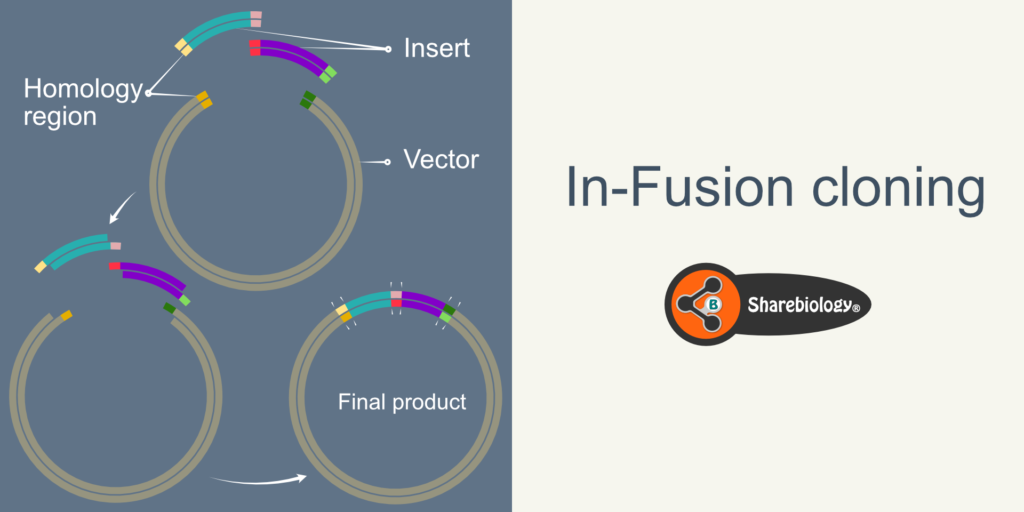
In-Fusion cloning or In-Fusion assembly is a ligation-free and directional molecular cloning method to clone one or multiple DNA fragments in any linearized vector in a single step and is a single-tube reaction. In conventional cloning, the presence and the availability of unique restriction enzyme sites in vectors and inserts limit the cloning. However, in In-Fusion cloning, the joining of DNA fragments depends upon the short stretch of homology (20 bp) between neighboring fragment ends. The neighboring fragments can be of different inserts (e.g., Gene and fluorescent protein) or insert and vector. The short stretch of homology between neighboring fragments will be incorporated in the primers used for PCR amplification of these fragments (inserts).
In-fusion cloning is Exonuclease-based cloning that uses the vaccinia virus’s DNA polymerase’s 3′ to 5′ exonuclease activity to generate single-stranded 5′ overhangs. The complementary single standard overhangs anneal together resulting in the joining of fragments (Fig 1.2 and 1.3).
Aslanidis and deJong originally reported the exonuclease-based technique in 1990. They used the (3′-5′) exonuclease activity of T4 DNA polymerase and successfully cloned the Alu1 fragments from human cosmid clones.
However, the exonuclease activity must be controlled to prevent the complete digestion of the DNA strand. To do so, the authors took advantage of the switching ability of T4 DNA polymerase’s polymerase and exonuclease activity. T4 DNA polymerase in the absence of nucleotides/dNTPs acts as an exonuclease and switches to polymerase function when provided with dNTPs. By exerting control over the exonuclease activity, the desired length of the overhangs can be achieved. The authors used 12-nucleotide overhangs at 5′ end of the primers to amplify the insert (Fig 1.1). These 12-nucleotide-overhangs designed lack Cytosine (C). After PCR amplification, the PCR products have 12 extra-base pairs (homology to the vector ends) and lack Guanine (G) in the complementary strand at the 3’end.
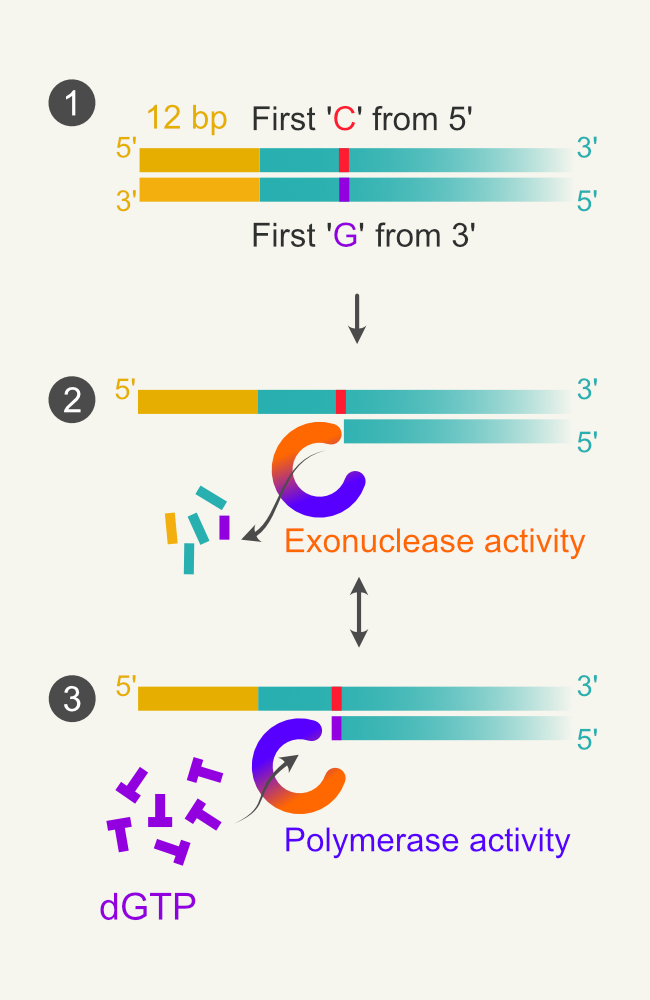
Figure 1. Illustration of Aslanidis and deJong’s experiment. 1.1 amplicon with 12 bp overhang and the position first encountered C:G pair from 5′ end. 1.2 T4 DNA polymerase switches to exonuclease activity and digests the 3′ ends of DNA. Small fragments represent dNMPs. 1.3 when the polymerase encounters C in the template strand, the polymerase switches to polymerase activity as the reaction mix contains only dGTP.
The PCR fragments are incubated with T4 DNA polymerase with a buffer containing only dGTP. Since the other dNTPs are absent, the exonuclease activity will remove the bases from the 3′ end until it encounters Guanosine (Fig 1.2). As the reaction mixture contains dGTP (only), the exonuclease activity will get switched to polymerase activity when it reads cytosine in the template strand. As the reaction buffer lacks other dNTPs, the enzyme cannot perform polymerization after that (Fig 1.3). This results in fragments with 5′ single-stranded tails with defined length (12 bases in this case + until it encounters G). The plasmid is also amplified using similar methodology used in the cloning.
However, T4 DNA polymerase requires specific controlled conditions, as mentioned above, to generate a specific length of single-stranded DNA. It also requires the addition of specific dNTP to stop the exonuclease reaction at which the enzyme is intended to stop the exonuclease activity.
In further research different enzymes and modifications were used to overcome the constraints posed by T4 DNA polymerase. Two such notable examples are the use of exonuclease III by Hsiao et al., 1993 and the use of T7 Gene6 exonuclease by Zhou and Hatahet 1995. However, these enzymes do have drawbacks. The exonuclease III is not useful for cloning DNA fragments with 3′-overhangs. The T7 Gene6 exonuclease-based cloning requires non-standard PCR primers (modified nucleotides, i.e., phosphorothioate) positioned towards the center (where the enzyme is intended to stop exonuclease activity) of the primers. The presence of phosphorothioate in primers simplifies the cloning reaction by avoiding the use of dNTPs to stop the exonuclease reaction at specific locations.
The In-Fusion cloning method does not require controlled conditions such as addition of dNTP, or the use of non-standard PCR primers.
Principle
In-Fusion cloning is a simple and efficient method of cloning that depends upon a 15 or 20 bp homology sequence. The 15 bp homology is sufficient for a successful In-Fusion reaction. however, increasing the size of homology sequence to 20 bp increases the cloning efficiency (Fig 2. Pairs of Yellow and red bars).
The In-Fusion cloning method uses 3′-5′ exonuclease activity of the vaccinia virus’s DNA polymerase. The 3′-5′ exonuclease removes the nucleotides at the 3′ end of the DNA, resulting in 5′ overhangs duplex (Fig 2.2). The complementary 5′ overhangs of duplex DNA fragments anneal together (insert-insert or insert-vector) making duplex–duplex recombinant intermediates (Fig 2.3). In-Fusion reaction mixture contains single-stranded DNA binding proteins that bind to any ssDNA outside the complementary region, enhancing transient complex stability (Fig 2.4).
The vaccinia virus exonuclease has a lesser affinity to nicked duplex DNA than to exposed ends, favoring more recombinant DNA molecules in the reaction. The nicks are unrepaired and sealed after entering E.coli via transformation using bacterial repair systems (Fig 2.5 A-C). Unlike other exonucleases, the In-Fusion enzyme mix does not require controlled conditions to make single-stranded overhangs (in length).
The In-Fusion method can be used to clone multiple fragments using a similar principle that was used to clone one fragment of DNA. Figure 3 illustrates the cloning of two fragments into one vector.
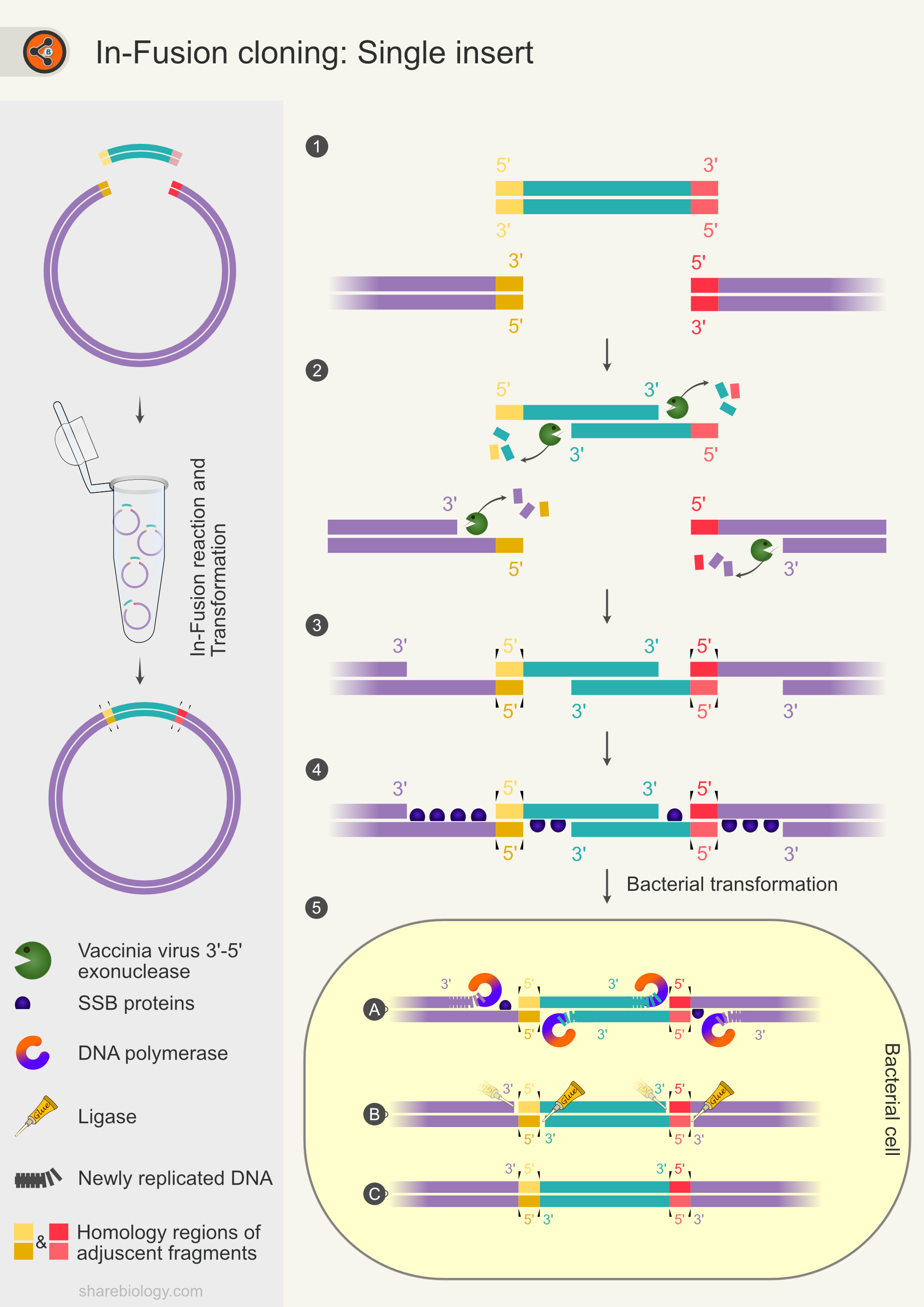
Figure 2. Schematic diagram representing steps in In-Fusion cloning for a single insert. The side panel represents the overview of In-Fusion cloning with one insert. 2.1 Illustration of vector and insert with 15-20 bp overlapping sequences. 2.2 Digestion of 3′ of insert and vector by vaccinia viral exonuclease activity, leaving 5′ overhangs. 2.3 Formation of the insert-vector duplex at the complementary sequences (ends of 5′ overhangs). The duplex is stabilized by hydrogen bonds between the complementary base pairs. 2.4 Single-stranded DNA is protected by single-stranded DNA binding proteins. 2.5 Transformation of the complex into E.coli. 2.5A-C DNA repair machinery fills the gaps and seals the nicks resulting in a covalently closed plasmid.
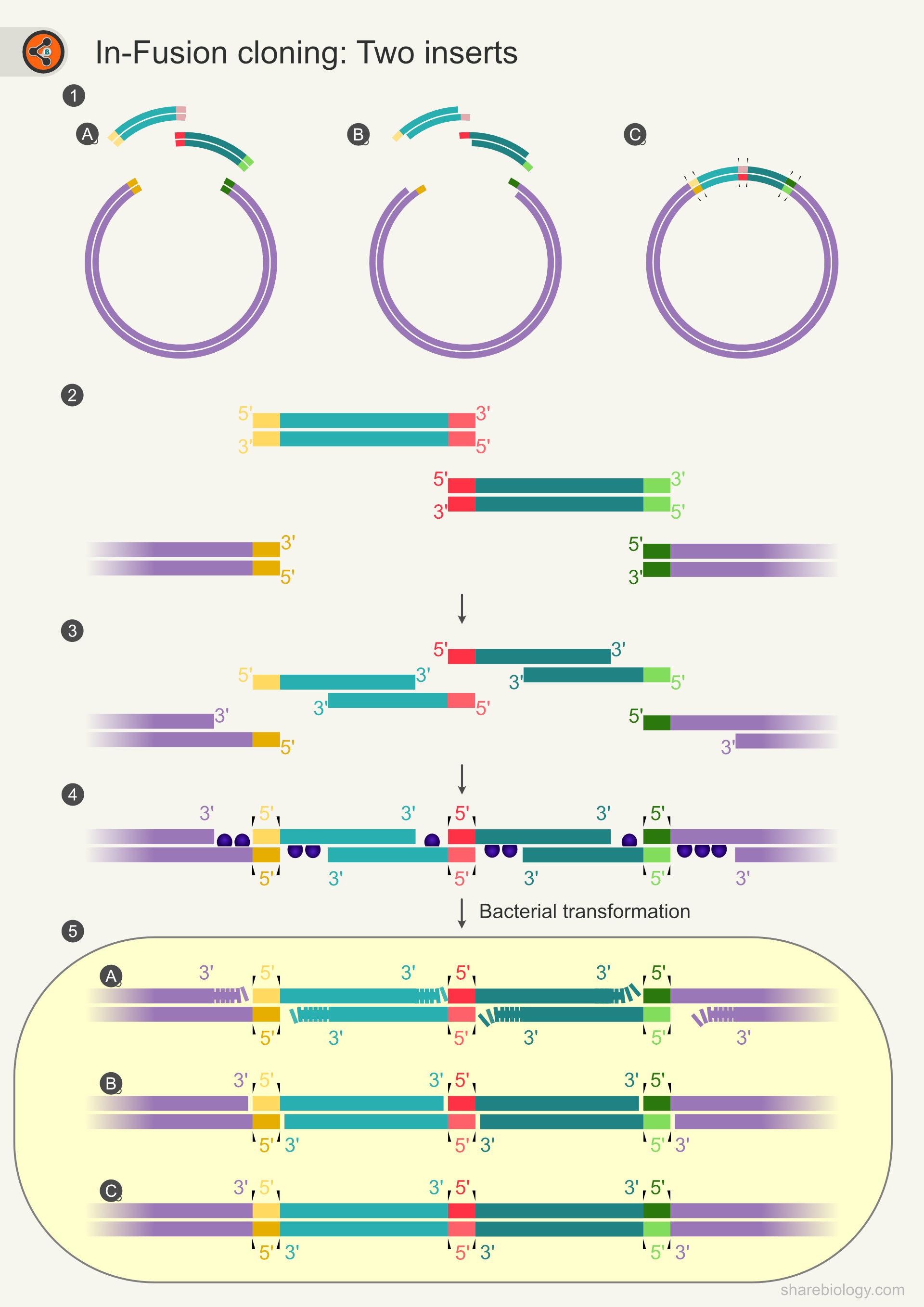
Figure 3: Schematic diagram representing In-Fusion cloning for two inserts.
Advantages
- DNA Ligase independent cloning
- Directional cloning with one or multiple DNA fragments cloned into any vector.
- Clone any insert, into any location, within any vector you choose as long as 20 bp overlap exists.
- It does not require restriction digestion (except vector linearization) or phosphatase treatment of vector as carried out in blunt-end cloning to prevent self-ligation.
- Unlike gateway cloning, this is seamless cloning i,e., it does not add or delete nucleotides.
- This method can be used for domain swaps, deletion, and insertion of sequences.
Limitations
- Extra care is required while designing the primer.
- The efficiency of In-Fusion cloning gets decreased by increasing the length and number of fragments in a reaction. However, this principle applies to almost all cloning methods.
- Without doing transformation, we cannot check the insert’s integrity or correct order (in multiple fragment cloning). It is not possible to confirm the integrity of the clone before the transformation through PCR as the fragments are not ligated.
- It might not be the right choice if the insert number is too high.
Procedure – an overview
Blunt-end cloning involves the following steps.
- Preparation of Insert
- Preparation of vector
- In-Fusion cloning reaction
- Transformation
- Selection of transformants and other downstream processes.
Let us see the first three steps in detail.
Preparation of Insert
The inserts for In-Fusion cloning will be amplified using In-Fusion-specific primers, which contains 20 bp homology with adjacent DNA fragments to which it will be joined. The adjacent DNA can be other insert DNA molecules or the plasmid backbone.
Preparation of vector (selecting a suitable vector and linearization)
The successful In-Fusion reaction requires a linearized vector. Select the suitable vector and linearize it using one of the following methods.
- The circular plasmid can be linearized using restriction enzymes which produce blunt ends or cohesive ends. You do not have to worry about the ends generated by the restriction enzymes as vaccinia exonuclease works on all types of ends (blunt, 3′ and 5′ overhangs).
- Amplification of the entire vector backbone using proofreading polymerase (Inverse PCR).
In-Fusion cloning reaction
The linearized plasmid/vector and insert(s) are mixed with a 2X In-Fusion HD enzyme reaction mixture and incubated at 50°C for 15 mins. After Keeping the samples on ice for 10-15 mins, the samples are will be ready for bacterial transformation.
Transformation
Transform the E.coli with 2.5 ul of In-Fusion reaction sample.
Detailed transformation protocol can be found here.
References
- In-fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations.
- Ligation-independent cloning of PCR products (LIC-PCR).
- Exonuclease III induced ligase-free directional subcloning of PCR products.
- An improved ligase-free method for directional subcloning of PCR amplified DNA.
- Takara.